Tomorrow, on 30 July 2026, the “GKV-Beitragssatzstabilisierungsgesetz” (GKV-BStabG) will enter into force, marking the culmination of a fast-paced and politically contentious legislative process. The reform, which has attracted significant attention, introduces a broad package of cost-containment measures across the statutory health insurance (GKV) system. Several of these specifically target pharmaceutical
Continue Reading What does the GKV-BStabG Reform Change for Pharma Pricing & Reimbursement in Germany? And the Reforms Are Not Over: What to Expect From the New German Minister of Health and the Pharma Dialogue?The Numbers Are In: What Financing has the Nagoya Protocol Delivered for Biodiversity After a Decade?
Bart Van Vooren and Yuliya Gevrenova
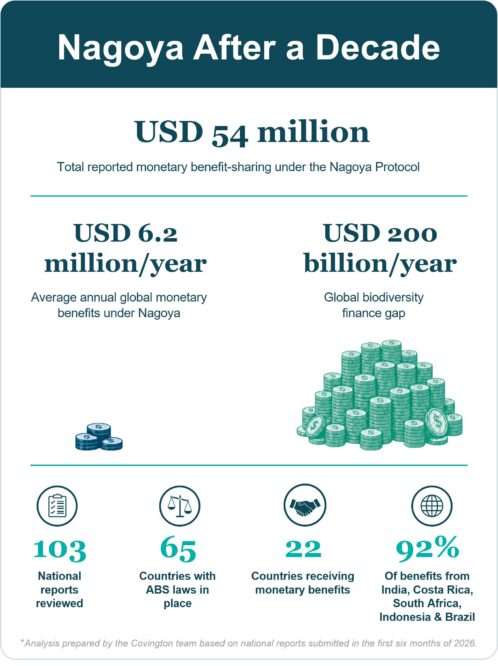
The Nagoya Protocol was adopted under the Convention on Biological Diversity (“CBD”) and is built on the idea that the commercial use of materials from nature (“genetic resources”) should translate into fair and equitable benefits for countries providing and protecting nature in their jurisdictions.
Continue Reading The Numbers Are In: What Financing has the Nagoya Protocol Delivered for Biodiversity After a Decade?Brazil delays SISGEN 3.0 Foreign-User Functionality while Government Advances alternative pathway for foreign ABS Compliance

Anderson Ribeiro, Aline Ferreira and Henryk Trelinski of Souto Correa Advogados contributed to the preparation of this article.
Foreign companies have long faced a practical challenge under Brazil’s access and benefit-sharing (“ABS”) regime: although registration obligations apply to activities involving Brazilian genetic heritage, foreign legal entities still cannot register
Continue Reading Brazil delays SISGEN 3.0 Foreign-User Functionality while Government Advances alternative pathway for foreign ABS ComplianceEU Biotech Act II: One Week Remaining to Shape the Commission’s Approach

The European Commission’s Call for Evidence on the upcoming Biotech Act II is open until 10 June 2026, with one week remaining for companies to provide input. Companies should consider engaging now to help shape the outcome.
The initiative is expected to have a broad impact across sectors including
Continue Reading EU Biotech Act II: One Week Remaining to Shape the Commission’s ApproachThe World Health Organization’s Emerging Pathogen Access System: Implications for Commercial Infectious Disease R&D

Over the past week, at the 79th World Health Assembly (“WHA”) in Geneva, governments have been debating one of the most consequential but still poorly understood elements of the World Health Organization (“WHO”) Pandemic Agreement that was adopted in May 2025: the Pathogen Access and Benefit-Sharing (“PABS”) System. Since
European Commission Publishes “ESRS 2.0” for Public Consultation: Draft Closely Follows EFRAG’s Technical Advice with Additional Simplifications for Companies
The European Commission (“Commission”) has launched a four-week feedback period — open until June 3, 2026 — on a draft delegated act to revise the European Sustainability Reporting Standards (“ESRS 2.0”). Ultimately, EU companies in scope of the EU’s Corporate Sustainability Reporting Directive (“CSRD”) will have to draft their annual
Continue Reading European Commission Publishes “ESRS 2.0” for Public Consultation: Draft Closely Follows EFRAG’s Technical Advice with Additional Simplifications for Companies
U.S.–UK Pharmaceutical Pricing Agreement: Learnings from the Published Text
On 2 April 2026, the UK Government published text memorializing its agreement with the U.S. in relation to pharmaceutical pricing. The UK now refers to this as an “arrangement” (“U.S.-UK Pharmaceutical Pricing Arrangement”). Our blog post from December discussed the in-principle heads of terms for the deal announced
Continue Reading U.S.–UK Pharmaceutical Pricing Agreement: Learnings from the Published TextGermany plans significant cuts in drug pricing and reimbursement – How would the GKV-Beitragssatzstabilisierungsgesetz impact pharmaceutical companies?
I. Background – From Pharma-Dialogue to Compulsory Price Cuts
Tomorrow, on 29 April 2026, the German Government is expected to adopt a new law to stabilize the finances of the statutory health insurances. This draft law titled “GKV-Beitragssatzstabilisierungsgesetz” (GKV-BStabG) proposes manifold cost-containment measures that would also significantly impact pharmaceutical companies.
Continue Reading Germany plans significant cuts in drug pricing and reimbursement – How would the GKV-Beitragssatzstabilisierungsgesetz impact pharmaceutical companies?Brazil declares Açaí a National Fruit: What Are the Real Practical Implications for EU Companies?

On January 8, 2026, Brazil published Law 15,330/2026, officially recognizing açaí berry as a Brazilian national fruit in a bid to protect it from so-called “biopiracy”, i.e., the illegal exploitation of genetic resources and traditional knowledge (“ATK”). Açaí berry is a ‘superfood’ rich in nutrients which grows almost
Navigating the new UN High Seas Treaty: Key Compliance Risks for Life Sciences Companies

On January 17th, 2026, the Biodiversity Beyond National Jurisdiction (“BBNJ”) Agreement, also known as the “High Seas Treaty”, entered into force. For the first time, companies that use marine genetic resources (“MGRs”) and digital sequence information (“DSI”) originating from areas beyond national jurisdiction may be required
Continue Reading Navigating the new UN High Seas Treaty: Key Compliance Risks for Life Sciences Companies